 Dan Serie
Dan SerieIt’s the late 2010’s, and genomic technologies have matured. The knowledge gleaned from yesterday’s basic research is beginning to make inroads into clinical practice, so it’s natural to seek out new frontiers. The central dogma of biology dictates that DNA stores our genetic information, RNA transcribes it for translation into proteins, and it’s these proteins which carry out the functions of life. But the story does not end there, as proteins themselves are decorated with various molecules that alter their function – so-called post-translational modifications (PTMs). Exacting measurement of these states will help unlock the full power of proteomics, i.e. the next frontier.
The most prominently studied PTMs, such as methylation, acetylation, and phosphorylation, are relatively straightforward. They consist of a handful of atoms and function as on-off switches – they’re either present at a given site, or not. Downstream effects of these binary PTMs have been illuminated in the study of epigenetics, histones, and protein kinases. The true wilderness is a little further out, in the realm of complex PTMs such as glycosylation.
A glycan is a branching tree of sugar chains, often consisting of hundreds of atoms, whose sequence and composition determine its structure and function. They are attached to asparagine (N-linked) or serine/threonine residues (O-linked) in translated peptide sequences. Glycans of various motifs can be found on membrane proteins in every cell in your body and enable a multitude of additional functions in secreted proteins. From facilitating intercellular communication to mediating interactions with the immune system, aberrant patterns of glycosylation have been implicated in all manner of disease [1-3]. Indeed, a coherent claim can be made for their evolutionary import [4]. But apart from a handful of well-characterized proteins (immunoglobulins in particular [5]), the heuristics by which they modify function largely remain unknown. This is mostly due to issues in assessment; compared to binary PTMs it’s significantly more difficult to measure long, branching oligosaccharides that can make up a substantial proportion of a proteoform’s weight.
But modern mass spectrometry has opened up new vistas of biological inquiry. Large scale measurement of site-specific glycosylation has only been possible in recent years, and advances in instrumentation and software have facilitated workflows that were previously tedious or impossible. The ability to reliably assess these states will enable downstream elucidation of biological function at a scale we could only dream of before. We stand on the cusp of an “epiproteomic” revolution, in which knowledge of fully specific proteoforms will supplant the hazy capabilities currently associated with a given protein (which are typically, as it stands, a weighted average of their possible states).
This is where InterVenn comes in. Our technology allows us to quantify the abundance of proteins and their site-specific glycosylation, promising great insight into the underlying biology of disease. We have just begun to mark associations between these widely varying glycoforms and their effect on human health. And indeed the number of possible glycoforms is staggering – the multiplicative expansion is huge for any PTM, but with the variety of glycan motifs available, the scale becomes astronomical.
Just what sort of scale are we dealing with? Prominent groups have attempted to address this question, through methods ranging from pure experimentation [6] to mathematical approaches [7]. Though the “correct” answer is currently out of reach, we can find approximations for several common PTMs. Here we proceed to calculate a rough, back-of-the-envelope estimate of the expansion of the proteome based on modifications taken one at a time
To arrive at such an estimate, we work with genome-wide averages. We calculate the mean number of available amino acid residues for each PTM from the UniProt reference sequence (UP000005640 [8]), pull the approximate percentage of those sites that are occupied from the literature, and couple this with the number of possible states per PTM (plus one for a non-modified state). From here an exponential equation yields the multiplicative modifier for the proteome (Figure 1).
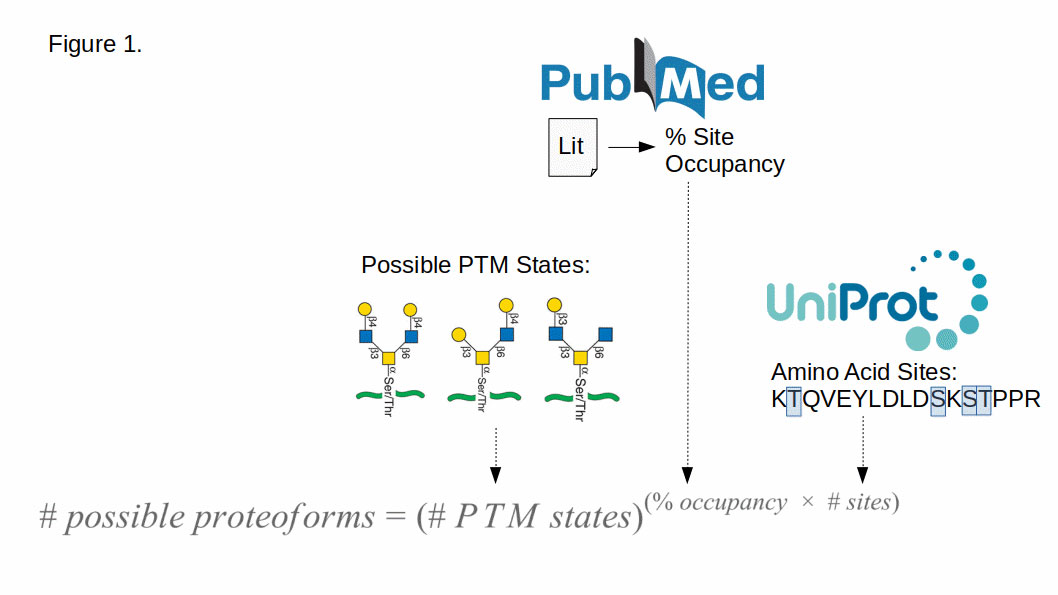
Of course, in doing so we have made a number of simplifying (possibly horrifying!) assumptions. For a complete rundown, see the appendix, but to wit: we ignore the contribution of protein isoforms, amino acid polymorphisms, and combinatorics generated by multiple simultaneous PTMs, we maximize the possible sites of attachment, ignore rare sites of modifications, and base site-occupancy on a literature review. Please check out the fine print.
With those caveats out of the way, we present the back of the envelope.

Generally speaking, the mathematical interplay between the number of potential sites, percent occupancy, and possible states favors PTMs with complex forms, i.e. glycosylation and ubiquitination. Phosphorylation has the largest number of possible sites, but still doesn’t come close to making up this difference in base. The low rate of acetylation may reflect the historical study of epigenetic modification of histones, though this role has been expanded upon further research [17].
O-linked glycosylation claims the biggest multiplier given its high number of sites, occupancy, and selection of motifs available. One can play with these numbers a bit, but qualitatively the complexity of the glyco-code remains dominant. Notably, N-linked glycosylation is one order of magnitude smaller than O-linked, though it’s likely that the larger N-glycans individually result in more structural and functional variation [18]. Interestingly, this point can be seen amidst the mathematics, where the selective residue pattern of N-glycans (with less than three sites per protein on average) and extraordinarily high site occupancy point towards the same deeper meaning: when and where N-linked glycosylation occurs, it’s vital. Recent studies assessing the complex interplay between glycan heterogeneity and protein dynamics [19] have borne out this conclusion.
Combining these estimates and multiplying by the number of genes in the database (20,874), we estimate a total of 4.19 billion possible proteoforms, based on singular PTM expansions alone. Without even considering PTM combinatorics, this is roughly twice the number of stars in the Milky Way! An obvious counterpoint is that the number of possible proteoforms is much greater than the number of existing proteoforms, and this is certainly true. But then, our colleagues working in the synthetic biology space may have something to say about that before too long…
At InterVenn we’re excited to tap into this new level of -omics, and hope to enable the broader research community to do the same. Changes in protein glycosylation are extraordinarily important, and understanding them will be necessary to unlock the power of proteomics. One danger is that this new language is too dense and ad hoc to yield to human understanding. Luckily, we also sit on the cusp of a golden era in deep learning research, and neural nets will serve us well as translators, interpreters, and enablers. As we set these technologies loose to make sense of this galaxy of molecular interactions, our goal remains applications in the greater multiverse of human health.
The Fine Print
References
2 Tower Place, 5th Floor,
South San Francisco, CA 94080
Bay Area Lab